
2月13日、14日,中国科学院分子植物科学卓越创新中心联合相关科研团队,分别在国际权威学术期刊《自然》和《细胞》上在线发表重要进展。
2025年2月14日,中国科学院分子植物科学卓越创新中心辰山科学研究中心陈晓亚院士团队与中国科学院遗传与发育生物学研究所高彩霞团队等合作在cell上在线发表了题为“design of coq10 crops based on evolutionary history(基于植物进化的辅酶q10性状设计)”的科研论文。该研究通过系统分析辅酶q在陆生植物中的演化轨迹及关键酶自然变异,解析了植物辅酶q侧链长度控制的分子机制,利用引导编辑技术改变水稻基因组coq1酶的5个氨基酸,创制了合成辅酶q10的水稻新种质,小麦编辑也取得重要进展。
2025年2月13日,中国科学院分子植物科学卓越创新中心万里研究组与张余研究组合作在nature上在线发表了题为“activation and inhibition mechanisms of a plant helper nlr”的研究论文。该研究揭示了植物免疫反应中通过形成异源三聚体激活与抑制细胞死亡的分子机制。
下面请看具体报道
《cell》研究成果:联合创制辅酶q10水稻新种质
辅酶q10与人体健康,尤其是心脏健康息息相关,它是线粒体呼吸链的电子传递体,也是脂溶性抗氧化剂。不同物种合成的辅酶q侧链长度不同,人体自身合成辅酶q10,侧链由10个异戊二烯单元(c50)组成,而水稻等谷物以及一些蔬菜和水果,主要合成辅酶q9,侧链含有9个异戊二烯单元(c45)。创制辅酶q10作物,提高植物食品中辅酶q10的含量,是一种性价比高且环境友好的营养强化新方法,意义重大。
为什么不同物种合成的辅酶q侧链长度不同,其分子机制一直不明。得益于上海辰山植物园丰富的植物资源,该团队采集了包括苔藓、石松、蕨类、裸子植物和被子植物在内的共67个科134种植物样品。检测各物种辅酶q类型及系统分布特征,发现辅酶q10是被子植物的祖先性状,多数植物仍然合成辅酶q10,而禾本科、菊科和葫芦科植物等主要合成辅酶q9。
要精准改造农作物性状,创造高营养品质,首先要精确锚定性状形成的关键因子。结合对1000多种陆生植物辅酶q侧链合成酶coq1氨基酸序列的进化分析和机器学习,科研团队最终确定了决定链长的5个氨基酸位点。通过精准编辑,创制了主要合成辅酶q10的水稻,其叶片和籽粒中辅酶q10占总辅酶q的75%,籽粒中辅酶q10达5 μg/g,且对水稻产量没有影响。基因编辑已成为一种高效安全的先进作物改良技术,编辑的植物不含外源基因、遗传稳定,近年来在发达国家发展迅速。q10水稻的研制成功,将大大丰富辅酶q10的食物来源,也为大数据和ai辅助育种提供了一个范例。
上海辰山植物园许晶晶副研究员、遗传发育所博士生雷源、上海交通大学张晓凡博士、分子植物卓越中心辰山科学研究中心李建戌副研究员为共同第一作者,分子植物卓越中心辰山科学研究中心陈晓亚院士和遗传发育所高彩霞研究员为共同通讯作者。该研究得到国家自然科学基金委员会、中国科学院、上海市科学技术委员会、上海市绿化和市容管理局辰山专项、云南省科技厅、新基石等项目的资助。
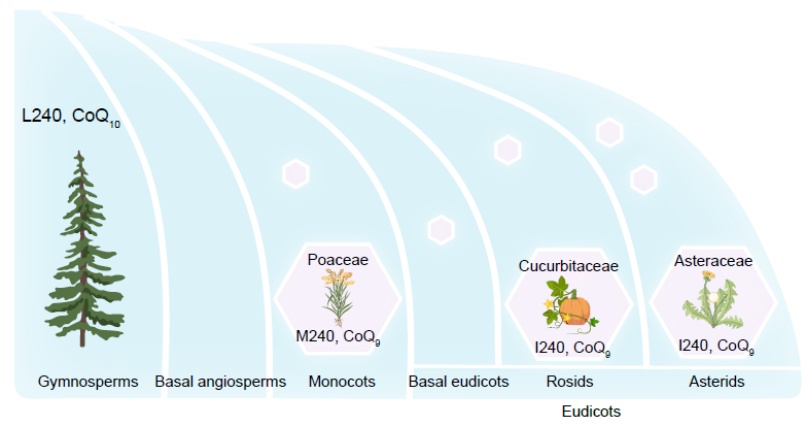
图1. 植物辅酶q种类的进化和关联氨基酸
图2. 引导编辑创制q10水稻
图3. 陈晓亚院士(右二)在田间观察水稻新种质
《nature》研究成果:在植物免疫机制上取得新突破
在漫长的生命历程中,植物为了抵御病原体的威胁,发展出了一套复杂的免疫防御系统。其中,细胞表面的模式识别受体 (pattern recognition receptors, prrs) 识别病原体相关分子模式,激活pti (pattern-triggered immunity)。细胞内的核苷酸结合富亮氨酸重复受体 (nucleotide-binding leucine-rich repeat receptors, nlrs) 识别病原体分泌的效应因子,激活eti (effector-triggered immunity)。根据n端结构域的不同,nlrs可分为tir-nlr (tnl)、cc-nlr (cnl) 和ccr-nlr (rnl) 三类。
其中,tnl和cnl作为sensor 感知病原菌效应因子并启动免疫信号传导;rnl则作为helper nlr,在sensor tnl的下游执行免疫功能。rnl分为adr1和nrg1两类。研究表明,tnl信号通路的激活依赖于三个关键的脂肪酶样蛋白eds1、pad4和sag101。当效应因子激活tnl时,产生的小分子信号诱导eds1-pad4与adr1结合,eds1-sag101与nrg1结合,并最终触发adr1和nrg1在质膜上寡聚化形成抗病小体,分别激活免疫抗性和细胞死亡通路。
在拟南芥中,全长的nrg1a和nrg1b作为正调控因子介导细胞死亡,且功能上冗余。n-端截短的nrg1c也能与eds1-sag101结合但负调控tnl介导的细胞死亡。eds1-sag101-nrg1a/b和eds1-sag101-nrg1c两种三元复合体形成的分子结构基础及其调控细胞死亡的分子机制不清楚。此外,eds1-sag101-nrg1a/b异源三聚体与nrg1a/b寡聚体之间的关系也有待深入研究。

本研究首先研究了tnl被激活后eds1-sag101-nrg1a异源三聚体形成与nrg1a寡聚体形成之间的关系。在tnl弱激活条件下,eds1-sag101-nrg1a异源三聚体形成明显,但nrg1a寡聚体形成不明显。在tnl强激活条件下,eds1-sag101-nrg1a异源三聚体形成不明显,但nrg1a寡聚体形成明显。这就暗示eds1-sag101-nrg1a异源三聚体与nrg1a寡聚体可能代表激活过程中不同的状态。
进一步测试发现在tnl被激活后,nrg1a寡聚化能力丧失的突变体 l134e能稳定eds1-sag101-nrg1a异源三聚体的形成。因此eds1-sag101-nrg1a三元复合体应该是代表nrg1a寡聚化前的中间状态。也就是说tnl激活产生的信号小分子导致eds1-sag101-nrg1a三元复合体的形成,但应该不足以引起nrg1a的寡聚化。
由于无法纯化得到eds1-sag101-nrg1a三元复合体的蛋白,研究团队重点关注了eds1-sag101-nrg1a l134e 这样一个更稳定的三元复合体并获得了高质量蛋白用于冷冻电镜结构解析。在eds1-sag101-nrg1a l134e三维结构中可以观察到tnl激活产生的信号小分子adrr-atp。adrr-atp结合在eds1-sag101中间并诱导sag101构象变化以促进eds1-sag101跟nrg1a l134e的互作。尽管研究团队用了全长的nrg1a l134e用于结构分析,但在eds1-sag101-nrg1a l134e三维结构中仅能观测到nrg1a l134e c端的whd-lrr部分,这表明nrg1a l134e的n端部分柔性较大且不参与互作。研究团队进一步在野生型nrg1a的背景下验证了在eds1-sag101-nrg1a l134e三维结构中观察的互作界面对tnl激活后的细胞死亡表型以及nrg1a寡聚化的重要性。
有趣的是,nrg1c仅含有whd-lrr结构域,正好是在eds1-sag101-nrg1a l134e结构中观察到的nrg1a参与互作的部分,这暗示拟南芥进化出nrg1c这样一个片段来进行负调控。通过对eds1-sag101-nrg1c冷冻电镜结构的分析,研究团队发现其与eds1-sag101-nrg1a l134e的结合方式高度相似,但nrg1c与eds1-sag101之间存在一些额外的相互作用导致nrg1c对eds1-sag101的亲和力更强,从而有效劫持eds1-sag101并抑制nrg1a激活的细胞死亡。研究团队之前的研究发现nrg1a通过其n端的ccr结构域中的正电氨基酸与细胞膜上带负电荷的磷脂互作定位于细胞膜并在细胞膜上寡聚化形成抗病小体。这里研究团队发现nrg1c由于缺失了n端的ccr而体现出核质分布,这样nrg1c就与eds1-sag101亚细胞定位完全一致从而实现更高效的劫持和负调控 (图a)。
eds1-sag101-nrg1是所有双子叶植物中保守存在的调控tnl介导细胞死亡的核心元件。团队的研究首次报道了eds1-sag101-nrg1的三维结构,并解析了其激活和抑制细胞死亡的多层精细调控的分子机理,极大推进了人们对这一重要免疫通路的认知。此外,研究团队发现nrg1与eds1-sag101的相互作用界面存在保守和非保守区域,这为解释不同植物物种中相关蛋白的不兼容性提供了理论依据,也为植物抗病设计育种提供了新思路。西湖大学柴继杰教授研究组在同期nature也报道了eds1-sag101-nrg1的三维结构,与团队的研究结果相互印证。
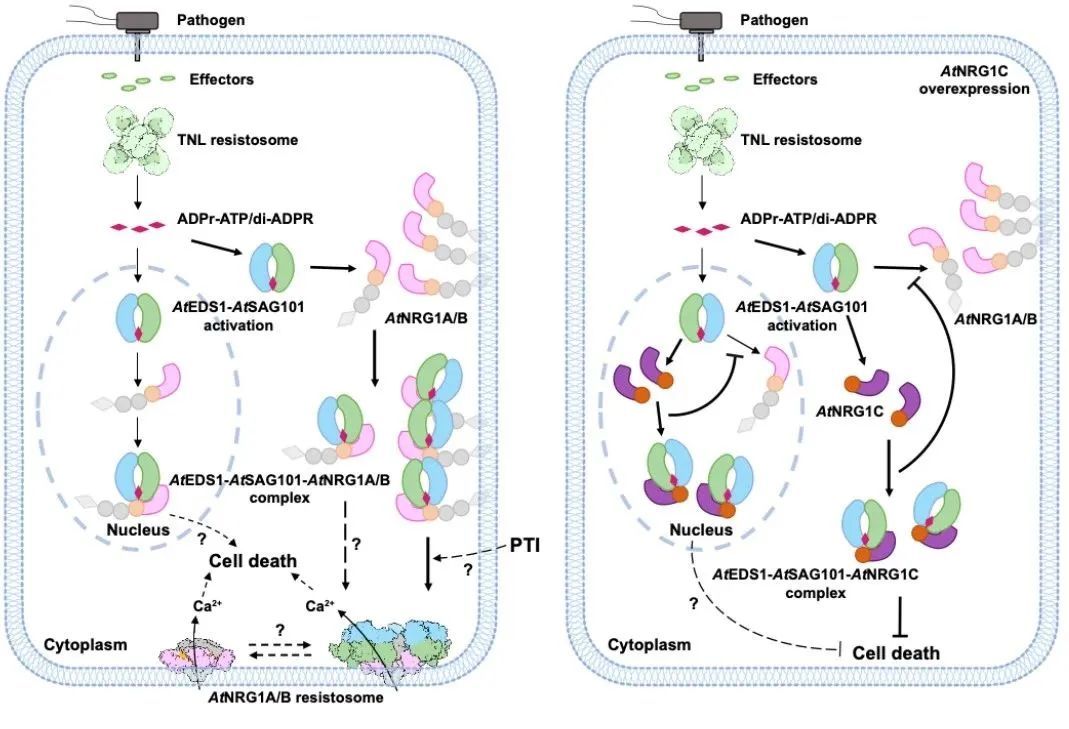
图a. eds1-sag101-nrg1三元复合体精细调控细胞死亡的模型
中国科学院分子植物科学卓越创新中心博士研究生肖银燕、副研究员武霄仙和博士后王再青为本文第一作者,万里研究员和张余研究员为本文通讯作者。博士研究生姬珂欣和赵杨研究员参与了该研究。该研究工作得到了国家重点研发计划、植物性状形成与塑造全国重点实验室、国家自然科学基金面上项目、中国科学院先导项目和上海市基础研究特区等经费资助。
来源:中国科学院分子植物科学卓越创新中心
编辑:蓝悦
↓分享
↓点赞
↓在看
上观号作者:上海科技